江蘇吉泰肽業科技有限公司
聯 系 人 :秦經理
座 機 :0515-84130555
手 機:17705109088
郵 箱:info@gtaipeptide.com
地 址:江蘇省鹽城市濱海縣新安大道799號
網 址:cn-cat.com.cn
郵 編:224500
1. 訂書肽發展簡介
訂書肽是基于多肽需形成α-螺旋通過細胞膜進入細胞的需求上發展起來的。生物體內的多種生命進程調節都是通過蛋白質與蛋白質之間的相互作用來實現的。例如病毒的自組裝,細胞的生長,分裂,分化等過程。而通常蛋白-蛋白相互作用的界面太大,從而使小分子藥物很難對其進行靶向定位,達到高效特異性地阻斷這種相互作用,展現良好的治療效果。蛋白類藥物因為很難順利通過細胞膜所以也達不到直接靶向細胞內相互作用的效果,因此,研究者們開始尋求一種能夠克服這兩種藥物缺點的既能夠進入細胞膜又能特異性靶向蛋白-蛋白相互作用的新的藥物分子。
研究表明,具有α-螺旋結構和富含正電荷的多肽可以穿過細胞膜。因此,人們開發了利用二硫鍵與分子內酰胺鍵作為支架的α-螺旋結構,但是,這些支架在生理環境下均不能穩定存在。2000年,Verdine等發展了一種用碳碳鍵作為支架來穩定多肽α-螺旋結構的方法,由此方法得到的多肽成為訂書肽(Stapled
peptides)。訂書肽有更高的α-螺旋程度,結合能力強,能通過細胞膜,難被蛋白酶水解,在生物體內半衰期長等優點。
2. 訂書肽合成
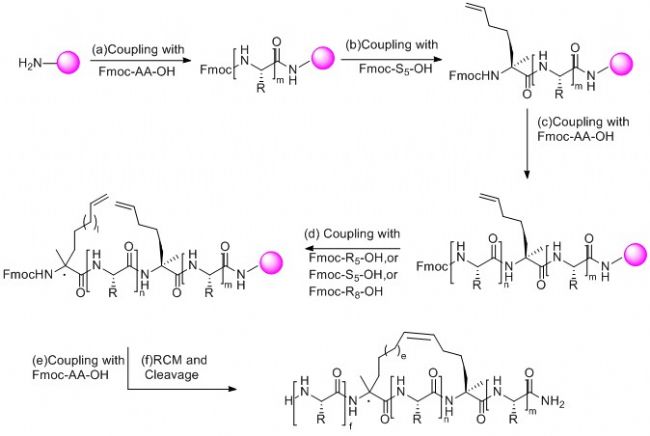
訂書肽合成策略為在固相合成肽鏈過程中引入兩個含有α-甲基,α-烯基的非天然氨基酸,然后兩個非天然氨基酸之間發生烯烴復分解反應(RCM)環化構成穩定α-螺旋結構構象的全碳支架,從而合成訂書肽。
1) α-甲基,α-烯基的非天然氨基酸的合成:
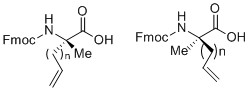
α-甲基,α-烯基的非天然氨基酸的一般結構
通常,在三乙胺的催化下,手性輔基試劑(1R,2S)-2-氨基-1,2-二苯乙醇與2-溴丙酸乙酯發生親核取代反應,并采用(Boc)2O保護自由胺基;接下來,在對甲苯磺酸的催化下,環狀的含有手性輔基的丙氨酸前體得以高效生產,在堿性催化劑和低溫下,烯基基團高效立體選擇性的連接到氨基酸的α-位,最后,在氨基鋰條件下脫去手性輔基,并采用Fmoc保護α-胺基。

α-甲基,α-烯基的非天然氨基酸的合成路線
2) 訂書肽的合成:
通常采用Fmoc-固相多肽合成方法制備訂書肽,兩個用于構象鎖定的α-甲基,α-烯基的非天然氨基酸之間一般間隔兩個,三個或者六個氨基酸,多肽合成完畢后,采用釕作催化劑進行烯烴復分解反應(RCM),如果實驗需要,還可以進行生物素(Biotin),熒光染色等修飾,最后,將目標多肽從樹脂上切割下來進行純化得到成品。